Scrna seq چیست
بخش اول – مبانی scRNA-seq
-
مقدمهای بر زیستشناسی تکسلولی – مفاهیم، اهمیت، و ظهور روشهای تکسلولی.
-
تکامل تاریخی توالییابی RNA – از Bulk RNA-seq تا تفکیک در سطح تکسلول.
-
اصول scRNA-seq – پایههای مولکولی و محاسباتی.
-
سؤالات زیستی که توسط scRNA-seq پاسخ داده میشوند – ناهمگونی بیان ژن، ردیابی دودمان سلولی (Lineage Tracing)، شناسایی انواع نادر سلول.
بخش دوم – طراحی آزمایش
-
آمادهسازی نمونه – جداسازی بافت، بررسی زندهمانی سلولها، آمادهسازی هسته در مقابل سلول کامل.
-
روشهای جداسازی سلول – FACS، میکروفلوئیدیک، کپسولهسازی قطرهای، و پلتفرمهای میکروول (Microwell).
-
گیراندازی RNA و رونویسی معکوس – مبانی مولکولی آمادهسازی کتابخانه scRNA-seq.
-
ساخت کتابخانه و پلتفرمهای توالییابی – Illumina، Nanopore، PacBio در کاربردهای تکسلولی.
بخش سوم – فناوریها و پروتکلها
-
روشهای مبتنی بر پلیت (Plate-Based) – Smart-seq، Smart-seq2، Smart-seq3.
-
روشهای مبتنی بر قطره (Droplet-Based) – 10x Genomics Chromium، Drop-seq، inDrop.
-
روشهای مبتنی بر میکروول (Microwell-Based) – Seq-Well، Microwell-seq.
-
پروتکلهای تخصصی – توالییابی RNA تکهستهای (snRNA-seq)، ترنسکریپتومیکس فضایی (Spatial Transcriptomics).
بخش چهارم – پردازش داده
-
جریانهای پیشپردازش (Preprocessing Pipelines) – پردازش بارکد سلولی، شناسههای مولکولی یکتا (UMIs).
-
کنترل کیفیت و فیلتر کردن – بررسی محتوای میتوکندری، شناسایی دابلتها، حذف RNA محیطی.
-
نرمالسازی و مقیاسبندی – رویکردهای آماری برای تنظیم دادهها.
-
کاهش ابعاد (Dimensionality Reduction) – PCA، t-SNE، UMAP.
-
خوشهبندی و برچسبگذاری نوع سلول – روشهای نظارتی و بدون نظارت.
بخش پنجم – کاربردهای زیستی
-
زیستشناسی تکوینی – درختهای دودمانی، تصمیمگیری سرنوشت سلولی، جنینزایی.
-
تحقیقات سرطان – ناهمگونی تومور، نقشهبرداری از میکروحیط ایمنی تومور.
-
علوم اعصاب – اطلس سلولهای مغزی، تنوع نورونی.
-
ایمنیشناسی – پروفایلبرداری از مجموعه گیرندههای ایمنی (Immune Repertoire)، تکوین سلولهای T و B.
-
پزشکی بازساختی – شناسایی سلولهای بنیادی، مدلسازی ارگانوئیدها.
بخش ششم – روشهای پیشرفته و یکپارچه
-
چنداُمیک (Multi-Omics) با scRNA-seq – CITE-seq، ادغام با scATAC-seq، چنداُمیک فضایی.
-
استنباط مسیر و آنالیز زمان کاذب (Pseudotime Analysis) – Monocle، Slingshot، PAGA.
-
تحلیل ارتباط سلول-سلول – جفتسازی لیگاند–گیرنده (Ligand–Receptor Pairing).
-
بازسازی شبکههای تنظیمی ژن – بررسی فعالیت فاکتورهای رونویسی.
بخش هفتم – چالشها و چشمانداز آینده
-
محدودیتها و سوگیریهای فنی – افت دادهها (Dropouts)، نویز ناشی از تقویت.
-
ذخیرهسازی داده و نیازهای محاسباتی – چالشهای دادههای حجیم.
-
استانداردسازی و قابلیت بازتولید – پروتکلها و دستورالعملهای گزارشدهی.
-
روندهای نوظهور در scRNA-seq – توان عملیاتی فوقالعاده بالا، توالییابی در محل (In Situ Sequencing)، کاربردهای بالینی.
مطالعه زیستشناسی در سطح تکسلول یکی از تحولآفرینترین تغییرات در علوم زیستی مدرن به شمار میرود. در روشهای سنتی زیستشناسی مولکولی و تحقیقات ترنسکریپتومیک، معمولاً تحلیلها بهصورت انبوه (Bulk) انجام میشود؛ به این معنی که RNA، DNA و پروتئینها از جمعیتهای بزرگی از سلولها استخراج و بهطور کلی اندازهگیری میشوند. هرچند این رویکردها بینشهای بزرگی در مورد فرایندهای سلولی فراهم کردهاند، اما بهطور ذاتی تفاوتهای میان سلولهای منفرد را پنهان میکنند و سیگنالهای ترنسکریپتومی را به یک پروفایل میانگین و همگن تبدیل میکنند.
این میانگینگیری میتواند پدیدههای زیستی مهم را پنهان کند، بهخصوص زمانی که انواع سلولهای نادر یا حالتهای گذرای سلولی وجود داشته باشد. زیستشناسی تکسلولی با هدف رفع این محدودیت پدید آمده و این امکان را فراهم میکند که ویژگیهای مولکولی در هر سلول بهصورت جداگانه اندازهگیری شود و تصویر دقیقتر و واضحتری از ناهمگونی سلولی ارائه دهد.
انگیزه اصلی برای مطالعه تکسلولی از این شناخت ناشی میشود که حتی سلولهای ژنتیکی یکسان میتوانند تفاوتهای چشمگیری در بیان ژنها، مقدار پروتئینها، فعالیت متابولیکی و رفتار عملکردی داشته باشند. این تفاوتها ممکن است به دلیل نوسانات تصادفی در رونویسی ژن، تغییرات اپیژنتیکی، تقسیمهای نامتقارن سلول، سیگنالهای محیطی و شبکههای سیگنالدهی پیچیده ایجاد شوند.
در جانداران پرسلولی، این تفاوت بین سلولها صرفاً نویز یا خطا نیست، بلکه اغلب نقش مهمی در فرایندهای زیستی ایفا میکند، مانند تمایز سلولی، پاسخهای ایمنی، حفظ تعادل بافتی و پیشرفت بیماریها.
به عنوان مثال:
-
در سرطان، یک زیرجمعیت کوچک از سلولها ممکن است الگوهای بیان ژنی داشته باشند که مقاومت دارویی ایجاد میکند و شناسایی زودهنگام آنها برای طراحی درمانهای مؤثر حیاتی است.
-
در زیستشناسی تکوینی، سلولهای پیشساز نادری با برنامههای رونویسی خاص ممکن است مسئول شکلگیری بافتهای تخصصی باشند.
زیستشناسی تکسلولی مجموعهای وسیع از راهبردهای آزمایشی و محاسباتی را شامل میشود که هدفشان دریافت و تحلیل اطلاعات مولکولی از هر سلول بهصورت جداگانه است. این حوزه شامل:
-
ترنسکریپتومیکس (بررسی مقدار mRNA برای ارزیابی فعالیت ژنها)
-
ژنومیکس (مطالعه تغییرات توالی DNA و ویژگیهای ساختاری)
-
اپیژنومیکس (بررسی دسترسی کروماتین و تغییرات DNA)
-
پروتئومیکس (اندازهگیری مقدار و تغییرات پروتئینها)
در میان این رویکردها، توالییابی RNA تکسلولی (scRNA-seq) به عنوان پرکاربردترین و قدرتمندترین ابزار برای درک پویایی بیان ژنها در مقیاس سلولی مطرح است. با امکان گرفتن و توالییابی ترنسکریپتوم هزاران تا میلیونها سلول بهصورت همزمان، این فناوری به محققان اجازه میدهد:
-
انواع سلولی را شناسایی کنند
-
مسیرهای تکوینی را استنباط کنند
-
پایههای مولکولی شرایط فیزیولوژیک و پاتولوژیک را کشف کنند
ظهور روشهای تکسلولی حاصل پیشرفتهای همزمان در چند حوزه است:
-
زیستشناسی مولکولی
-
میکروفلوئیدیک (امکان جداسازی سلولها در قطرات یا میکروولهای نانولیتر)
-
توالییابی پرتوان (High-throughput Sequencing) که حالا میتواند میلیونها خوانش کوتاه را با سرعت و هزینه پایین تولید کند
-
زیستمحاسبات که الگوریتمهای تخصصی برای نرمالسازی دادهها، کاهش ابعاد، خوشهبندی و استنباط مسیر سلولی توسعه داده است
این پیشرفتها باعث شدهاند که ترنسکریپتومیکس تکسلولی از پروژههای کوچک و اکتشافی به تلاشهای بزرگ مقیاس برای ساخت اطلس سلولی کل بافتها و حتی موجودات کامل تبدیل شود.
اهمیت زیستشناسی تکسلولی فراتر از تحقیقات پایه رفته و به کاربردهای ترجمهای و بالینی رسیده است:
-
در انکولوژی، این روش تنوع جمعیتهای سلولی تومور، سلولهای استرومال و سلولهای ایمنی نفوذکننده را آشکار کرده است.
-
در ایمنیشناسی، این فناوری تنوع سلولهای T و B را روشن کرده و درک بهتری از پاسخهای ایمنی و خودایمنی فراهم آورده است.
-
در علوم اعصاب، زیرگروههای مولکولی نورونها و سلولهای گلیال شناسایی شده و اصول جدیدی از سازماندهی و عملکرد مغز کشف شده است.
همچنین ادغام ترنسکریپتومیکس تکسلولی با ترنسکریپتومیکس فضایی (که موقعیت مکانی سلولها را در بافت حفظ میکند) بینش بیسابقهای از تعاملات سلولی در محیطهای طبیعیشان فراهم کرده است.
بهطور کلی، زیستشناسی تکسلولی یک تغییر پارادایم در نحوه نگاه زیستشناسان به سیستمهای زنده است. به جای فرض یکنواختی در جمعیتهای سلولی، این حوزه ناهمگونی را یک ویژگی ذاتی و آموزنده در نظر میگیرد. مطالعه سلولها به صورت منفرد، مدلهای دقیقتر و مکانیکیتری از تکوین، فیزیولوژی و بیماریها ارائه میدهد.
با این حال، این تغییر نگرش چالشهای جدیدی ایجاد کرده است، از جمله:
-
تفسیر دادههای پرنویز و پراکنده
-
مقیاسپذیری آزمایشها برای میلیونها سلول
-
ادغام دادههای ترنسکریپتومی با سایر انواع دادههای مولکولی
در ادامه این مرجع، ابتدا پیشینه تاریخی منجر به توسعه scRNA-seq بررسی خواهد شد و سپس به اصول، روششناسیها و کاربردهای این فناوری تحولآفرین پرداخته میشود.
فصل ۲ – تکامل تاریخی توالییابی RNA
تاریخچهٔ توالییابی RNA (RNA-seq) داستانی از کنجکاوی علمی، نوآوری فناورانه و تلاش بیوقفه برای درک بنیانهای مولکولی حیات است. پیش از ظهور فناوریهای توالییابی با توان بالا، دانشمندان از روشهایی مانند نورترن بلاتینگ (Northern blotting)، واکنش زنجیرهای پلیمراز با رونویسی معکوس (RT-PCR) و میکروآرایهها (Microarrays) برای سنجش میزان RNA استفاده میکردند.
این روشهای اولیه گرچه اطلاعات ارزشمندی ارائه میدادند، اما در گستره، حساسیت و انعطافپذیری محدود بودند:
-
نورترن بلاتینگ که در اواخر دههٔ ۱۹۷۰ توسعه یافت، امکان شناسایی مولکولهای RNA خاص را فراهم میکرد اما فرآیندی پرزحمت و کمبازده داشت.
-
RT-PCR که در اواخر دههٔ ۱۹۸۰ معرفی شد، حساسیت و توان کمیسازی را بهبود بخشید، ولی همچنان روشی هدفمند بود و نیاز به دانش قبلی از توالیهای مورد اندازهگیری داشت.
-
میکروآرایهها که در میانهٔ دههٔ ۱۹۹۰ پدید آمدند، امکان اندازهگیری همزمان هزاران رونویس را فراهم کردند، اما به دلیل اتکا به پروبهای از پیش تعریفشده، کشف ژنها یا واریانتهای جدید را محدود میکردند.
گذار به نسل جدید
با افزایش نیاز پژوهشگران به درک کامل تنوع و پویایی ترنسکریپتوم (مجموعهٔ کامل مولکولهای RNA در یک سلول یا بافت در یک زمان خاص)، محدودیتهای روشهای قدیمی آشکارتر شد.
پیشرفت در فناوریهای توالییابی DNA بهویژه ظهور پلتفرمهای نسل جدید توالییابی (NGS) در میانهٔ دههٔ ۲۰۰۰، تحول بزرگی ایجاد کرد. پلتفرمهایی مانند Illumina (توالییابی به روش سنتز) و SOLiD (توالییابی مبتنی بر اتصال) توان را به شکل چشمگیری افزایش داده و هزینهها را کاهش دادند، و امکان بررسی جامع و بدون سوگیری جمعیتهای RNA را فراهم کردند.
اولین مطالعات RNA-seq که حدود سال ۲۰۰۸ منتشر شدند نشان دادند که این روش نه تنها میتواند کمیت رونویسهای شناختهشده را اندازهگیری کند، بلکه قادر است ژنهای جدید، الترنتیو (Alternative splicing)، بیان وابسته به آلل و RNAهای غیرکدکننده را نیز شناسایی کند — ویژگیهایی که در روشهای مبتنی بر میکروآرایه قابل دستیابی نبودند.
مزایای بنیادین RNA-seq
RNA-seq با تبدیل RNA به cDNA و توالییابی آن در قالب میلیونها قطعهٔ کوتاه، این امکان را به پژوهشگران داد که تعداد خوانشهای همراستا با هر ژن را بهصورت دیجیتال بشمارند و بنابراین اندازهگیری کمی و دقیق از سطوح بیان ژن به دست آورند.
برخلاف روشهای هیبریداسیون، RNA-seq نیاز به دانش قبلی از توالیها نداشت و این امکان را فراهم کرد که ترنسکریپتهای ناشناخته در ژنومهای کمتر شناختهشده شناسایی شوند و مقایسهٔ ترنسکریپتوم بین گونهها انجام شود. همچنین، بهبود طول خوانش و استفاده از توالییابی دو سر (Paired-end) باعث شد اتصالهای پیچیده و تغییرات ساختاری بهتر شناسایی شوند.
تخصصیتر شدن پروتکلها
با بلوغ RNA-seq، پروتکلهای تخصصی برای پرسشهای خاص زیستی توسعه یافتند:
-
RNA-seq اختصاصی رشته برای تمایز بین رونویسهای سنس و انتی سنس.
-
حذف RNA ریبوزومی و انتخاب پلی(A) برای غنیسازی RNAهای کدکننده و غیرکدکننده موردنظر.
-
توالییابی هدفمند RNA برای تمرکز بر نواحی خاص ترنسکریپتوم.
-
توالییابی RNA کامل برای پروفایلکردن هم RNAهای کدکننده و هم غیرکدکننده.
همچنین پژوهشگران شروع به ادغام دادههای RNA-seq با اپیژنوم و پروتئوم کردند تا چارچوبهای چنداُمیک برای درک تنظیم بیان ژن بسازند.
محدودیتها و ظهور توالییابی تکسلولی
با وجود تمام این پیشرفتها، RNA-seq تودهای (Bulk) همچنان با مشکل میانگینگیری روی جمعیتهای بزرگ سلولی مواجه بود. این موضوع در بافتهای ناهمگن مانند تومورها، جنینهای در حال رشد یا اندامهای ایمنی مشکلساز بود، چون انواع سلولهای نادر و حیاتی ممکن بود زیر سیگنال سلولهای غالب پنهان شوند.
برای حل این مشکل، دانشمندان به سراغ پروفایلکردن ترنسکریپتوم در سطح تکسلول رفتند که مستلزم پیشرفت در جداسازی سلولها، به دام انداختن مولکولها، کوچکسازی واکنشها و افزایش حساسیت برای کار با مقدار بسیار کم RNA (چند پیکوگرم) بود.
اولین تلاشها در سالهای ۲۰۰۹ تا ۲۰۱۱ با روشهایی مانند Tang و همکاران که شامل جداسازی دستی سلول منفرد و سپس تقویت cDNA بود، انجام شد. این روشها کمبازده و پرزحمت بودند، اما نشان دادند که بازسازی ترنسکریپتوم یک سلول منفرد امکانپذیر است.
با پیشرفت میکروفلوئیدیک و سیستمهای قطرهای، scRNA-seq توانست به صورت موازی هزاران تا میلیونها سلول را پردازش کند. تا اواسط دههٔ ۲۰۱۰، پلتفرمهای تجاری مانند 10x Genomics Chromium و Fluidigm C1 این فناوری را در دسترس آزمایشگاههای سراسر جهان قرار دادند و عصر جدید زیستشناسی تکسلولی آغاز شد.
فراتر از یک تحول فنی
گذار از توالییابی تودهای به توالییابی RNA تکسلولی صرفاً یک پیشرفت فنی نبود، بلکه یک تغییر پارادایم در پژوهش زیستی محسوب میشود. RNA-seq بنیانی برای دیدگاههای جامع و کمی نسبت به بیان ژن ایجاد کرد، و scRNA-seq این توان را به وضوح تکسلولی گسترش داد تا ناهمگنی سلولی را آشکار کند.
درک این مسیر تاریخی برای قدردانی از تأثیر تحولآفرین scRNA-seq و شناخت اینکه چگونه درسهای آموختهشده از فناوریهای ترنسکریپتومیک پیشین هنوز بر روشهای تکسلولی اثر میگذارند، ضروری است.
فصل ۳ – اصول بنیادی توالییابی RNA تکسلولی (scRNA-seq)
توالییابی RNA تکسلولی (scRNA-seq) رویکردی تحولآفرین است که به پژوهشگران امکان میدهد بیان ژن را در سطح تکسلول بررسی کنند و پیچیدگی حالتها و عملکردهای سلولی را که معمولاً در اندازهگیریهای جمعی پنهان میمانند، آشکار سازند.
در اصل، scRNA-seq بر سه اصل اساسی استوار است:
-
جداسازی فیزیکی سلولها بهصورت منفرد.
-
جداسازی و تبدیل RNA هر سلول به DNA مکمل (cDNA).
-
توالییابی موازی و انبوه cDNA برای ایجاد پروفایلهای کمی ترنسکریپتومی.
هر یک از این مراحل به روشهای تخصصی نیاز دارند تا مقادیر بسیار اندک RNA از هر سلول حفظ، بهدرستی تکثیر، و با دقت در دادههای توالییابی بازنمایی شوند.
۱. جداسازی تکسلولها
هدف این است که ترنسکریپتوم هر سلول به طور مستقل اندازهگیری شود؛ بنابراین باید سلولها را از یک بافت پیچیده یا سوسپانسیون مخلوط سلولی، بدون آلودگی از سلولهای مجاور، جدا کرد. روشهای مختلفی برای این کار وجود دارد:
-
جداسازی دستی (مانند میکرومانیپولاسیون یا پیپت دهانی) که در مطالعات اولیه استفاده میشد اما کند، پرزحمت و کمظرفیت بود.
-
سیستمهای میکروفلوئیدیک که سلولها را با استفاده از کانالها و شیرهای کنترلشده به محفظههای واکنش هدایت میکنند.
-
روشهای مبتنی بر قطره (droplet-based) که هر سلول را در یک قطره نانولیتر حاوی مواد لیزکننده و آنزیمهای رونویسی معکوس محصور میکنند.
-
جداسازی به کمک فلوسایتومتری (FACS) یا میکروبرش لیزری (LCM) که دقت بالایی در انتخاب سلولهای خاص دارند، بهویژه برای سلولهای نادر یا زمانی که زمینه فضایی مهم است.
۲. جداسازی RNA و رونویسی معکوس (Reverse Transcription)
برخلاف DNA، مولکولهای RNA بسیار ناپایدار هستند و بهسرعت توسط آنزیمهای RNase تجزیه میشوند. بنابراین:
-
لیز سلول باید بهسرعت و در شرایط غیرفعالکننده RNase انجام شود تا RNA پایدار بماند.
-
RNA آزاد شده با آنزیم رونویسی معکوس به cDNA تبدیل میشود.
-
از آنجا که یک سلول تنها حدود ۱۰ تا ۵۰ پیکوگرم RNA دارد، این مرحله باید بسیار کارآمد باشد تا از دست رفتن اطلاعات به حداقل برسد.
-
بسته به هدف مطالعه، از پرایمرهای Oligo(dT) برای mRNAهای پلیآدنیله یا پرایمرهای هگزامر تصادفی برای پوشش گستردهتر کل ترنسکریپتوم استفاده میشود.
۳. تکثیر cDNA و توالییابی
به دلیل مقدار کم cDNA، تکثیر برای ایجاد ماده کافی جهت توالییابی ضروری است. روشها شامل:
-
PCR یا تکثیر خطی با رونویسی درونکشتگاهی (IVT) که هرکدام سوگیریها و خطاهای متفاوتی دارند.
-
استفاده از شناسههای مولکولی منحصربهفرد (UMI) که توالیهای تصادفی کوتاهی هستند و قبل از تکثیر به هر مولکول RNA متصل میشوند تا سوگیریهای تکثیر تصحیح و دقت کمی دادهها افزایش یابد.
-
در نهایت، cDNA تکثیرشده به کتابخانههای توالییابی تبدیل میشود که با پلتفرمهای پرظرفیت (مانند Illumina) سازگار هستند.
۴. بارکدگذاری سلولی (Cellular Barcoding)
یکی از مفاهیم کلیدی scRNA-seq بارکدگذاری سلولی است که اجازه میدهد هزاران سلول به طور همزمان پردازش و توالییابی شوند، در حالی که هویت ترنسکریپتوم هر سلول حفظ میشود.
در سیستمهای قطرهای، هر قطره حاوی مهرههایی با الیگونوکلئوتیدهای بارکددار است. با لیز سلول، RNAها به این پرایمرهای متصل به مهره متصل شده و همه ترنسکریپتهای یک سلول با یک بارکد خاص برچسبگذاری میشوند. پس از توالییابی، نرمافزارها از این بارکدها برای بازسازی پروفایل بیان ژن هر سلول استفاده میکنند.
۵. تحلیل محاسباتی
scRNA-seq فقط یک تکنیک آزمایشگاهی نیست، بلکه بخش عمدهای از آن تحلیلی و محاسباتی است:
-
دمولتیپلکس کردن دادهها بر اساس بارکد سلولی.
-
همترازی (Alignment) به ژنوم یا ترنسکریپتوم مرجع.
-
شمارش و کمیسازی خوانشها.
-
کنترل کیفیت و حذف نویزهای فنی.
-
استفاده از الگوریتمهای تخصصی برای نرمالسازی دادهها، حذف خطاهای فنی، خوشهبندی سلولها، تحلیل بیان افتراقی و بازسازی مسیرهای تمایز (Trajectory Inference).
جمعبندی
اصول بنیادی scRNA-seq بر پایهی:
-
دستکاری دقیق سلولها و RNA آنها
-
تبدیل کارآمد RNA به cDNA
-
تکثیر مقاوم در برابر خطا
-
تخصیص دقیق دادهها به هر سلول با بارکدگذاری
استوار است. این اصول حاصل دههها نوآوری در زیستشناسی مولکولی هستند که برای حساسیت بسیار بالای تحلیل تکسلولی تطبیق یافتهاند. تسلط بر این اصول، پژوهشگران را قادر میسازد تا مناظر ترنسکریپتومی پنهان در بافتها، اندامها و کل موجودات را آشکار کنند و دید بیسابقهای از تنوع و پویایی سلولی به دست آورند.
فصل ۴ – جریان کاری آزمایشگاهی توالییابی RNA تکسلولی (scRNA-seq)
جریان کاری آزمایشگاهی در توالییابی RNA تکسلولی یک زنجیره دقیق و هماهنگ از مراحل است که هر کدام باید بهینهسازی شوند تا نتایج دقیق، قابلتکرار و از نظر زیستی معتبر به دست آید.
از آنجا که scRNA-seq بیان ژن را در تفکیک تکسلولی اندازهگیری میکند، این فرآیند آزمایشگاهی با چالشهایی روبهروست که در RNA-seq کلی (bulk) وجود ندارند، از جمله:
-
مقدار ورودی بسیار کم ماده ژنتیکی
-
حساسیت بالا به آلودگی
-
لزوم حفظ وضعیت رونویسی سلولها از لحظه جداسازی
این جریان کاری بهطور کلی شامل مراحل زیر است:
۱. آمادهسازی نمونه و جداسازی سلولها
۲. جداسازی تکسلولی
۳. گیراندازی RNA و رونویسی معکوس
۴. تقویت (Amplification) cDNA
۵. آمادهسازی کتابخانه (Library preparation)
۶. توالییابی
۷. ارزیابی اولیه کیفیت دادهها
با وجود اصول یکسان، اجرای هر مرحله بسته به پلتفرم انتخابی (صفحهای، قطرهای، میکروول، یا فناوریهای جدیدتر) میتواند متفاوت باشد.
۱. آمادهسازی نمونه و تجزیه سلولها
اولین چالش، بهدستآوردن سوسپانسیون سلولهای منفرد و زنده از بافت یا نمونه اولیه است.
-
در بافتهای جامد مانند مغز، کبد یا نمونه تومور، نیاز به هضم آنزیمی (مثل کلاژناز، تریپسین، پاپائین) همراه با جداسازی مکانیکی ملایم (پیپتاژ یا صاف کردن) برای شکستن ماتریکس خارج سلولی وجود دارد.
-
مدت زمان هضم باید دقیق کنترل شود:
-
هضم بیشازحد → آسیب غشا و نشت RNA
-
هضم ناکافی → تشکیل توده سلولی و مشکل در جداسازی تکسلولی
-
-
در نمونههای مایع مثل خون، هضم کمتر نیاز است، اما ممکن است لیز گلبول قرمز یا جداسازی با گرادیان چگالی لازم باشد.
-
برای سلولهای نادر یا شکننده، باید مراقبت ویژه برای حفظ زندهمانی و تمامیت RNA انجام شود.
-
دما معمولاً پایین نگه داشته میشود (۴ درجه سانتیگراد) تا فعالیت متابولیک کند و تغییرات مصنوعی بیان ژن به حداقل برسد.
۲. جداسازی تکسلولی
بعد از تهیه سوسپانسیون، باید هر سلول جداگانه پردازش شود.
روشها:
-
FACS (فلوسایتومتری با جداسازی فلورسانس) → انتخاب سلول بر اساس اندازه، شکل و مارکرهای فلورسانس (مناسب برای غنیسازی جمعیتهای خاص).
-
میکرومانیپولاسیون دستی → مناسب برای سلولهای خاص یا بزرگ (کم throughput).
-
پلتفرمهای میکروفلوئیدیک (مثل 10x Genomics Chromium) → سلولها در قطرات نانولیتر همراه با دانههای بارکددار کپسوله میشوند.
-
سیستمهای میکروول → سلولها در چاهکهای کوچک همراه با پرایمرهای بارکددار گیر میافتند.
-
لیزر کپچر میکرودیسسکشن (LCM) → برداشت مستقیم سلولها از برش بافتی برای حفظ اطلاعات فضایی.
انتخاب روش بستگی به میزان throughput، اندازه سلول و نیاز به حفظ جمعیتهای نادر یا بافتی دارد.
۳. گیراندازی RNA و رونویسی معکوس
-
سلولها لیز میشوند تا RNA آزاد شود.
-
به دلیل آسیبپذیری RNA، بافر لیز حاوی مهارکننده RNase و دترجنت برای شکستن غشا است.
-
در روشهای قطرهای، لیز و رونویسی معکوس در همان قطره انجام میشود.
-
پرایمرهای بارکددار سه بخش اصلی دارند:
-
بارکد سلولی (برای تشخیص هویت سلول)
-
UMI (شناسه مولکولی منحصربهفرد برای هر RNA)
-
Oligo(dT) (برای اتصال به دم پلیآدنیلات mRNA)
-
-
آنزیم رونویس معکوس RNA را به cDNA تبدیل میکند.
-
کارایی این مرحله بسیار مهم است چون هر RNA که از دست برود، دیگر بازیابی نمیشود.
۴. تقویت (Amplification) cDNA
-
هر سلول پستاندار فقط چند ده پیکوگرم RNA دارد → تقویت ضروری است.
-
روشها:
-
PCR → سریع، ساده، اما با بایاس بیشتر (بهخصوص در توالیهای GC بالا یا طولانی).
-
IVT (رونویسی درونکشتگاهی) → یکنواختتر ولی زمانبرتر.
-
-
UMIها کمک میکنند تا بایاس تقویت در مرحله تحلیل داده اصلاح شود.
۵. آمادهسازی کتابخانه (Library Preparation)
-
تکهتکهکردن (Fragmentation) cDNA
-
اتصال آداپتورهای توالییابی (با ترانسپوزاز یا آنزیمها)
-
آداپتورها شامل توالیهای لازم برای اتصال به Flow Cell و آغاز واکنش توالییابی هستند.
-
اضافه کردن Index برای مولتیپلکس کردن نمونهها در یک اجرای توالییابی.
۶. توالییابی
-
بیشتر روی پلتفرم Illumina انجام میشود (دقت بالا، throughput زیاد، هزینه نسبتا پایین).
-
عمق توالییابی (Reads per cell) مهم است:
-
کمعمق (۲۰–۵۰ هزار read/سلول) → پوشش تنوع سلولی با هزینه کمتر
-
عمیق (>۱ میلیون read/سلول) → شناسایی دقیقتر رونویسیهای کمبیان و ایزوفرمها
-
-
طول خوانش: کوتاه (۵۰–۱۰۰ bp) برای بیان ژن کافی است، بلندتر برای تحلیل ایزوفرمها و ترنسکریپتهای فیوژن.
۷. ارزیابی اولیه کیفیت
-
بررسی طول خوانش، کیفیت بازها، آلودگی به آداپتور، و میزان داده تولیدی.
-
دمولتیپلکس کردن با استفاده از بارکدها برای اختصاص reads به سلولهای صحیح.
-
استفاده از UMI برای حذف تکرارهای PCR.
-
حذف سلولهای با:
-
تعداد خوانش کم
-
محتوای بالای RNA میتوکندریایی (نشانه آسیب یا مرگ سلول)
-
✅ جمعبندی: جریان کاری scRNA-seq یک زنجیره حساس و دقیق است که هر مرحلهاش بر کیفیت و تفسیر دادهها تأثیر مستقیم دارد. انتخاب روش هضم، پلتفرم جداسازی، راهبرد گیراندازی RNA، شیوه تقویت، و عمق توالییابی همه تعیینکننده کیفیت نهایی دادهها هستند. تسلط کامل بر هر مرحله = نقشهبرداری دقیق و معتبر از ترنسکریپتوم تکسلولها.
فصل ۵ – پردازش و تحلیل محاسباتی دادههای scRNA-seq
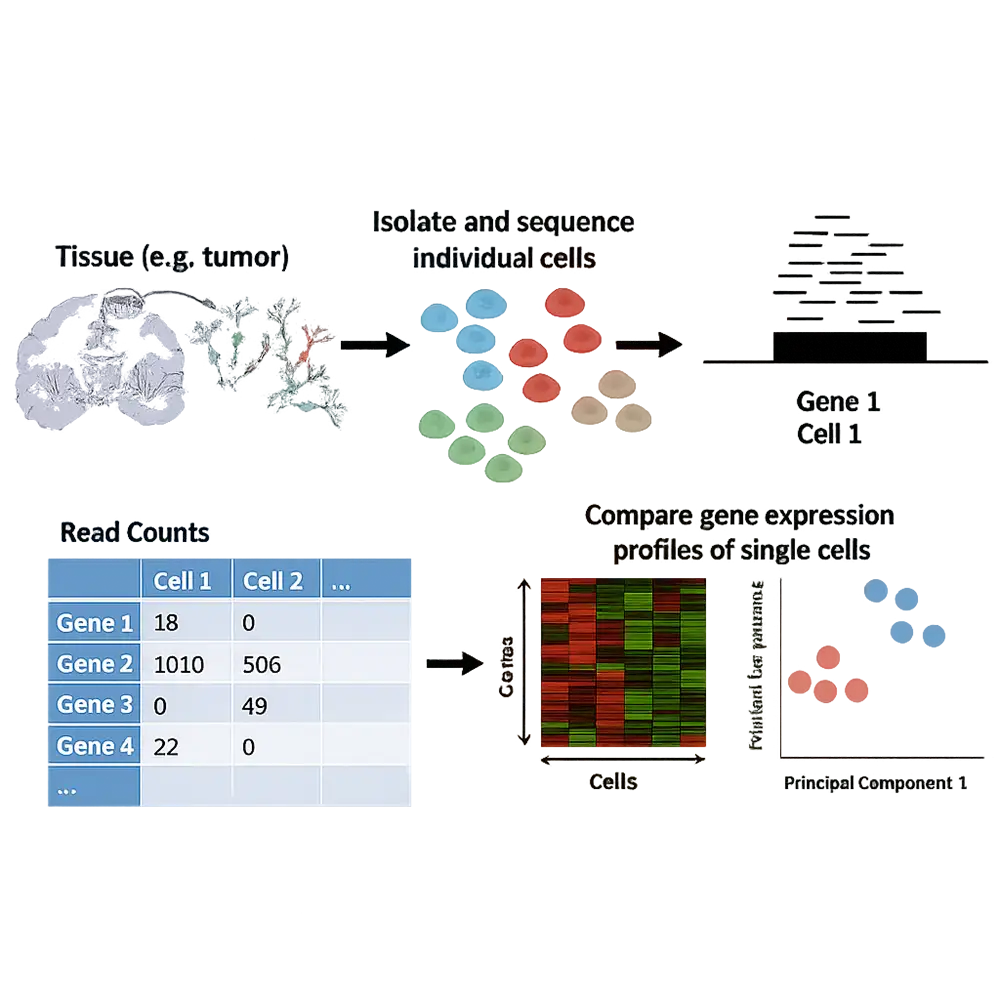
تحلیل محاسباتی دادههای توالییابی RNA تکسلولی (scRNA-seq) یک فرآیند چندمرحلهای است که دادههای خام حاصل از توالییابی را به بینشهای زیستی معنادار تبدیل میکند. از آنجا که scRNA-seq دادههایی با بُعد بالا و ویژگیهای نویزی خاص تولید میکند، باید جریان کاری تحلیلی بهدقت طراحی شود تا چالشهایی مانند تفاوتهای فنی (technical variability)، رخدادهای dropout و اثرات دستهای (batch effects) را برطرف کند. این فصل یک مرور جامع بر خط لوله پردازش و تحلیل محاسباتی ارائه میدهد که از پیشپردازش دادههای خام آغاز و به تفسیر پیشرفته نتایج ختم میشود.
کنترل کیفیت اولیه دادهها
مرحله نخست با کنترل کیفیت دادههای خام شروع میشود. دستگاههای توالییابی فایلهای FASTQ تولید میکنند که حاوی میلیونها read کوتاه هستند. هر read در پروتکلهای مبتنی بر UMI دارای یک بارکد سلولی و یک شناسه مولکولی یکتا (UMI) است.
ابزارهایی مانند FastQC برای ارزیابی کیفیت پایهها، میزان GC و آلودگی به آداپتورها استفاده میشوند.
-
Reads با کیفیت پایین (مثلاً با امتیاز Phred پایین) یا حذف یا اصلاح میشوند تا از ورود خطا به مراحل بعد جلوگیری شود.
-
توالیهای آداپتور با استفاده از ابزارهایی مثل Cutadapt یا Trimmomatic حذف میشوند.
-
همزمان، توالیهای UMI استخراج میشوند تا در مراحل بعدی، خوانشهای تکراری که از یک مولکول RNA مشترک ایجاد شدهاند، حذف (deduplication) شوند.
همترازی (Alignment) یا شبههمترازی (Pseudo-alignment)
پس از تمیزکردن reads، نوبت به همترازی میرسد:
-
ابزارهایی مانند STAR یا HISAT2، reads را به ژنوم یا ترنسکریپتوم مرجع نگاشت میکنند و شمارش دقیق بیان ژنها را ممکن میسازند.
-
ابزارهای شبههمترازی مانند Kallisto یا Salmon با سرعت بیشتر، reads را به ترنسکریپتهای سازگار متصل میکنند و از همترازی کامل صرفنظر میکنند.
در پروتکلهای مبتنی بر UMI، نرمافزارهایی مانند UMI-tools خوانشهایی که UMI و ژن یکسان دارند را ادغام میکنند تا شمارشها بهطور مصنوعی بالا نرود.
خروجی این مرحله یک ماتریس بیان ژن بهازای هر سلول است که پایه و اساس تمام تحلیلهای بعدی است.
کنترل کیفیت در سطح سلول
در این مرحله، سلولها بر اساس معیارهایی مانند:
-
تعداد کل UMI،
-
تعداد ژنهای شناسایی شده،
-
درصد خوانشهای میتوکندریایی،
فیلتر میشوند. -
سلولهایی با شمارش بسیار پایین احتمالاً قطرات خالی (empty droplets) هستند.
-
سلولهایی با شمارش غیرعادی بالا ممکن است دوبلت یا مالتیپلت باشند.
-
درصد بالای ژنهای میتوکندریایی نشانه سلولهای آسیبدیده یا در حال مرگ است و حذف میشوند.
همچنین ژنهایی که در تعداد بسیار کمی از سلولها بیان میشوند نیز حذف میگردند تا نویز کاهش یابد.
نرمالسازی دادهها
نرمالسازی برای اصلاح تفاوتهای عمق توالییابی و بهرهوری برداشت RNA انجام میشود:
-
روشهای ساده مثل مقیاسبندی بر اساس Counts Per Million (CPM)، تبدیل لگاریتمی و پایدارسازی واریانس، مقادیر را بین سلولها قابل مقایسه میکنند.
-
روشهای پیشرفته مانند SCTransform (در چارچوب Seurat) با مدلسازی رابطه میان میانگین و واریانس، نویز فنی را حذف کرده و سیگنالهای زیستی را حفظ میکنند.
اصلاح اثرات دستهای (Batch Effect Correction)
وقتی دادهها از چندین اجرای توالییابی یا شرایط آزمایشی مختلف بهدست میآیند، اثرات دستهای میتوانند سیگنالهای واقعی زیستی را مخدوش کنند.
-
روشهایی مانند Harmony، یکپارچهسازی Seurat یا MNN، ویژگیهای مشترک بین دستهها را شناسایی و دادهها را تنظیم میکنند.
-
احتیاط: تصحیح بیش از حد میتواند باعث حذف تفاوتهای واقعی زیستی شود.
کاهش بُعد دادهها
دادههای scRNA-seq بسیار پربُعد هستند:
-
PCA بهعنوان یک روش خطی، محورهای اصلی تغییرات را استخراج میکند.
-
روشهای غیرخطی مانند t-SNE و UMAP برای بصریسازی ساختار پیچیده جمعیت سلولی استفاده میشوند.
این روشها معمولاً روی خروجی PCA اعمال میشوند تا نویز و هزینه محاسبات کاهش یابد.
خوشهبندی (Clustering) و شناسایی زیرجمعیتهای سلولی
-
روشهای گرافمحور مثل Louvain یا Leiden یک گراف نزدیکترین همسایهها ایجاد میکنند و خوشهها را شناسایی میکنند.
-
پارامتر Resolution تعیین میکند خوشهبندی چقدر جزئی یا کلی باشد.
هر خوشه باید با اعتبارسنجی زیستی بررسی شود تا نشاندهنده یک نوع یا حالت سلولی واقعی باشد.
شناسایی ژنهای نشانگر (Marker Genes)
-
تحلیل بیان افتراقی (DE) بین خوشهها و سایر سلولها، ژنهایی که در یک خوشه بهطور خاص بیان بالایی دارند را مشخص میکند.
-
آزمونهای آماری مانند Wilcoxon rank-sum، Likelihood ratio یا مدلهای مبتنی بر آمار پیشرفته به کار میروند.
-
با استفاده از پایگاههای داده مرجع یا مطالعات پیشین، ژنهای نشانگر به انواع سلولی شناختهشده نگاشت میشوند.
تحلیل مسیر تکاملی و شبهزمان (Trajectory Inference & Pseudotime)
-
روشهایی مانند Monocle، Slingshot یا PAGA ترتیب سلولها را بر اساس زمان شبهزیستی بازسازی میکنند.
-
این کار روابط تبارزایی و حالات انتقالی را آشکار میکند.
-
کاربرد ویژه در زیستشناسی تکوینی و پزشکی بازساختی دارد.
روشهای پیشرفته و یکپارچهسازی چندوجهی
-
SCENIC فعالیت فاکتورهای رونویسی را پیشبینی میکند.
-
چارچوبهایی مانند Seurat v5 یا Liger دادههای scRNA-seq را با ATAC-seq تکسلولی، پروتئومیکس یا اپیژنومیک ادغام میکنند.
-
این کار تصویر جامعتری از حالت و عملکرد سلول ارائه میدهد.
بصریسازی و گزارشدهی
-
نمودارهای UMAP رنگبندیشده بر اساس نوع سلول
-
ویولنپلاتها (Violin plots) برای نمایش بیان ژنهای نشانگر
-
Heatmap ژنهای افتراقی
مستندسازی دقیق روشها، پارامترها و آستانههای QC برای قابلیت تکرارپذیری ضروری است.
جمعبندی
پردازش و تحلیل دادههای scRNA-seq یک جریان کاری پیچیده و حساس است که از خوانشهای خام شروع شده و تا تفسیر پیشرفته زیستی ادامه دارد. هر تصمیم در مراحل پیشپردازش، نرمالسازی، تصحیح دستهای، کاهش بُعد، خوشهبندی و شناسایی میتواند نتیجهگیری نهایی زیستی را تغییر دهد. بهترین روشها بر شفافیت، تکرارپذیری و ادغام با سایر دادههای چندوجهی تأکید دارند تا حداکثر توان scRNA-seq بهکار گرفته شود.
فصل ۶ – یکپارچهسازی scRNA-seq با سایر دادههای اُمیک و دادههای فضایی
یکپارچهسازی توالییابی RNA تکسلولی (scRNA-seq) با سایر دادههای اُمیک و مجموعه دادههای ترنسکریپتومیک فضایی، بهعنوان یک راهبرد قدرتمند برای دستیابی به درک جامعتر از حالتها و عملکردهای سلولی ظهور کرده است.
اگرچه scRNA-seq وضوح بینظیری از ناهمگونی ترنسکریپتومی ارائه میدهد، اما فاقد بستر مکانی است و بهطور مستقیم سایر لایههای تنظیمات زیستی را — مانند دسترسپذیری کروماتین، تغییرات اپیژنتیکی، میزان پروتئین یا ترکیب متابولیتی — اندازهگیری نمیکند.
ترکیب scRNA-seq با روشهای مکمل، این محدودیتها را برطرف کرده و امکان شناسایی چندبعدی سلولها در محیط طبیعیشان را فراهم میکند.
۱. یکپارچهسازی با اپیژنومیک تکسلولی
یکی از رایجترین گسترشهای scRNA-seq، یکپارچهسازی با اپیژنومیک تکسلولی است.
-
scATAC-seq میزان دسترسپذیری کروماتین را در مقیاس تکسلولی اندازهگیری میکند و دیدگاهی در مورد عناصر تنظیمی کنترلکننده بیان ژن ارائه میدهد.
-
با ارتباط دادن قلههای scATAC-seq به ژنهای نزدیک، پژوهشگران میتوانند ناحیههای کروماتینی باز را با فعالیتهای ترنسکریپتومی که توسط scRNA-seq سنجیده شدهاند، مرتبط کنند.
-
چارچوبهای محاسباتی مانند Seurat v5، Signac و Liger امکان تحلیل مشترک scRNA-seq و scATAC-seq را فراهم میکنند.
-
این رویکردها شناسایی فاکتورهای رونویسی، تعاملات تقویتکننده–پروموتر و برنامههای اپیژنتیکی تعریفکننده هویت سلول را ممکن میسازند.
۲. ترنسکریپتومیک فضایی
یکی از کمبودهای کلیدی scRNA-seq، از دست رفتن اطلاعات مکانی به دلیل تفکیک بافت است.
-
روشهایی مانند 10x Genomics Visium، Slide-seq و MERFISH بیان ژن را با حفظ معماری بافت ثبت میکنند.
-
ادغام با scRNA-seq معمولاً شامل استفاده از پروفایل ترنسکریپتومی با وضوح بالا برای تجزیه سیگنالهای ترکیبی دادههای فضایی است.
-
ابزارهایی مانند Seurat، SpaOTsc و Tangram میتوانند انواع سلولی شناساییشده توسط scRNA-seq را دوباره به موقعیت مکانیشان در بافت نگاشت کنند.
-
این رویکرد در مطالعه فرایندهای تکوینی، میکروحیطه تومور و بازسازی بافت ارزش ویژهای دارد، جایی که روابط مکانی برای درک عملکرد حیاتی هستند.
۳. یکپارچهسازی با پروتئومیک تکسلولی
افزودن پروتئومیک تکسلولی دامنه بیولوژیکی را با اندازهگیری مستقیم میزان پروتئین گسترش میدهد.
-
روشهایی مانند CITE-seq و REAP-seq با استفاده از تگهای آنتیبادی، همزمان پروتئینهای سطحی یا داخل سلولی و ترنسکریپتومها را میسنجند.
-
این اندازهگیری دوگانه مشکل همبستگی ناقص بین mRNA و پروتئین را برطرف میکند که ناشی از تنظیم پسارونویسی و پایداری پروتئین است.
-
چارچوبهای محاسباتی مانند TotalVI دادههای چندوجهی تکسلولی را یکپارچه و نویززدایی میکنند تا استنباط دقیقتر ممکن شود.
۴. چنداُمیکهای ترکیبی
برخی پلتفرمهای تجربی مانند SHARE-seq یا SNARE-seq امکان اندازهگیری همزمان RNA و دسترسپذیری کروماتین را از یک سلول واحد فراهم میکنند.
-
دیگر روشها، ترنسکریپتومیک را با متیلاسیون DNA یا پروفایل تغییرات هیستونی ترکیب میکنند.
-
ادغام این دادهها به نرمالسازی بینمودالیته، همترازی فضاهای نهفته و اصلاح سوگیریهای خاص هر روش نیاز دارد.
-
الگوریتمهایی مانند MOFA+ و نسخههای چنداُمیکی CCA منابع مشترک تغییرات را شناسایی کرده و ویژگیهای خاص هر مدالیته را حفظ میکنند.
۵. ادغام با متابولومیک و آزمونهای عملکردی
-
هرچند متابولومیک تکسلولی مستقیم از نظر فنی دشوار است، اما متابولومیک فضایی و اندازهگیریهای تودهای از جمعیتهای سلولی جداشده میتوانند با دادههای ترنسکریپتومی پیوند محاسباتی پیدا کنند.
-
این ادغام در سرطانشناسی برای ارتباط بازبرنامهریزی متابولیکی با زیرگونههای سلولی تومور و در ایمونولوژی برای بررسی تنظیم متابولیکی فعالسازی سلولهای ایمنی کاربرد دارد.
۶. هماهنگسازی دادهها و حذف اثرات بچ
یک چالش مهم در ادغام چنداُمیکی، هماهنگسازی دادههاست:
-
هر مدالیته ساختار نویزی و مقیاس اندازهگیری خاص خود را دارد.
-
الگوریتمهایی مانند همسایههای متقابل نزدیک، همترازی گراف و خودرمزگذارهای واریاسیونی برای نگاشت دادهها به یک نمایش نهفته مشترک به کار میروند.
-
کیفیت ادغام با بررسی همترازی انواع سلول شناختهشده بین مدالیتهها و حفظ الگوهای مشترک و اختصاصی سنجیده میشود.
۷. دستاوردهای علمی
-
در سرطانشناسی، ترکیب اطلاعات ترنسکریپتومی، اپیژنتیکی و فضایی باعث شناسایی زیرکلونهای تومور، مسیرهای تکاملی آنها و تعاملاتشان با سلولهای ایمنی و استرومایی شده است.
-
در زیستشناسی تکوینی، ادغام چنداُمیکی سلسلهمراتبهای تبارزایی را با وضوح بیسابقه بازسازی کرده است.
-
در ایمونولوژی، چنداُمیک تکسلولی فضایی سازمان فضایی زیرمجموعههای سلولهای ایمنی و شبکههای ارتباطیشان را آشکار کرده است.
۸. چشمانداز آینده
-
فناوریها به سمت تحلیل چندمقیاسی، همزمان و با توان عملیاتی بالا حرکت میکنند.
-
پیشرفتهای آینده امکان تحلیل میلیونها سلول در چندین مدالیته را فراهم میکنند.
-
این مسیر ما را به سوی دستیابی به یک اطلس سلولی کامل موجودات زنده پیش میبرد، که در آن هر سلول در چندین لایه تنظیمی و در جایگاه فضایی و عملکردی طبیعیاش توصیف شده است.
فصل ۷ – کاربردهای scRNA-seq در زیستشناسی تکوینی، ایمنیشناسی و پژوهشهای بیماری
توالییابی تکسلولی RNA یا scRNA-seq، درک ما از سامانههای پیچیده زیستی را با فراهم کردن امکان شناسایی و تحلیل ناهمگنی سلولی با جزئیاتی بیسابقه متحول کرده است. کاربردهای این فناوری گستره وسیعی از حوزهها را شامل میشود؛ از ترسیم مسیرهای تکوینی سلولها گرفته تا رمزگشایی پاسخهای ایمنی و مشخص کردن ویژگیهای بیماریها. در هر یک از این حوزهها، scRNA-seq اطلاعاتی ارائه میدهد که با روشهای سنتی bulk RNA-seq دستیافتنی نبود و نشان میدهد چگونه سلولهای منفرد در عملکرد، پاتولوژی و تکامل بافتها و ارگانها نقش دارند.
۱. زیستشناسی تکوینی (Developmental Biology)
در زیستشناسی تکوینی، scRNA-seq به ابزاری کلیدی برای بازسازی سلسلهمراتب دودمانی سلولها و درک رخدادهای مولکولی پشتصحنه تصمیمگیریهای سرنوشت سلولی تبدیل شده است.
-
با بررسی پروفایل ترنسکریپتومی هزاران سلول در بازههای زمانی مختلف رشد، پژوهشگران میتوانند مسیرهای تمایز سلولی را با استفاده از ابزارهای محاسباتی مانند Monocle، Slingshot و PAGA پیشبینی کنند.
-
این تحلیلها حالتهای سلولی گذار را آشکار میکنند که اغلب نقاط کلیدی تصمیمگیری در فرایند ارگانوژنز (تشکیل اندامها) هستند.
-
در مطالعات تکوین اولیه جنین، scRNA-seq جمعیتهای نادری از سلولهای پیشساز را شناسایی کرده و شبکههای تنظیم ژنی تعیینکننده هویت آنها را مشخص ساخته است.
-
در فرآیندهای تخصصی مانند نوروزنز (تشکیل سلولهای عصبی)، هماتوپوئیز (خونسازی) و کاردیوژنز (تشکیل قلب)، این فناوری نقشههای دقیقی از ارتباط فعالیت فاکتورهای رونویسی، مسیرهای پیامرسانی و تغییرات اپیژنتیکی تا ایجاد سلولهای تخصصی فراهم کرده است.
۲. ایمنیشناسی (Immunology)
در ایمنیشناسی، scRNA-seq نمایی جامع از پیچیدگی سیستم ایمنی ارائه میدهد و امکان شناسایی زیرجمعیتها و حالتهای عملکردی مختلف سلولهای ایمنی در سلامت و بیماری را فراهم میکند.
-
برخلاف فنوتیپگذاری ایمنی سنتی که بر مارکرهای از پیش تعیینشده تکیه دارد، scRNA-seq اجازه میدهد جمعیتهای ایمنی جدید و ناشناخته کشف شوند.
-
این روش در شناسایی فرسودگی سلولهای T در عفونتهای مزمن و سرطان، انواع ماکروفاژهای التهابی در بیماریهای خودایمنی و پویایی بلوغ سلولهای B در مراکز زایگر بسیار مؤثر بوده است.
-
ادغام scRNA-seq با توالییابی گیرندههای سلولهای T (TCR) و گیرندههای سلولهای B (BCR) امکان تحلیل همزمان ارتباطهای کلونال و وضعیتهای ترنسکریپتومی را فراهم کرده است که پلی میان ویژگی آنتیژنی و فنوتیپ عملکردی ایجاد میکند.
-
در تحقیقات واکسن، scRNA-seq برای ردیابی تکامل پاسخهای ایمنی در طول زمان استفاده شده و شاخصهای کلیدی حفاظت و حافظه ایمنی را مشخص کرده است.
۳. تحقیقات سرطان (Cancer Research)
کاربرد scRNA-seq در سرطانشناسی بسیار تحولآفرین بوده است.
-
تومورها ذاتاً ناهمگن هستند و از سلولهای سرطانی با پروفایلهای ژنتیکی، اپیژنتیکی و ترنسکریپتومی متفاوت، به همراه سلولهای غیرسرطانی بافت همبند و ایمنی تشکیل شدهاند.
-
scRNA-seq این پیچیدگی را با شناسایی زیرکلونهایی با فنوتیپهای تکثیر، تهاجم یا مقاومت دارویی متفاوت آشکار کرده است.
-
این فناوری برای مطالعه برهمکنشهای تومور–ایمنی حیاتی بوده و مکانیسمهای فرار ایمنی و جذب سلولهای سرکوبگر ایمنی مانند سلولهای T تنظیمی و سلولهای سرکوبگر مشتق از میلوئید را روشن ساخته است.
-
مطالعات طولی تکسلولی بر روی بیماران سرطانی نشان دادهاند که تومورها چگونه در پاسخ به درمانهای هدفمند، ایمنیدرمانی یا شیمیدرمانی سازگار میشوند و زیرجمعیتهای مقاوم ایجاد میکنند که بازگشت بیماری را سبب میشوند.
۴. سایر بیماریها
فراتر از سرطان و ایمنیشناسی، scRNA-seq در زمینههای گوناگونی کاربرد یافته است:
-
بیماریهای عصبیتحلیلبرنده (مانند آلزایمر، پارکینسون و ALS): شناسایی زیرگروههای جدید نورونی و حالتهای فعالسازی گلیا مرتبط با بیماری.
-
اختلالات متابولیک (مانند دیابت نوع ۲): شناسایی زیرجمعیتهای β-سلولی ناکارآمد و برنامههای پاسخ به استرس که باعث اختلال در ترشح انسولین میشوند.
-
بیماریهای قلبیعروقی: پروفایلسازی سلولهای اندوتلیال، عضله صاف و ایمنی در طول پیشرفت آترواسکلروز و بازسازی پس از سکته قلبی.
-
بیماریهای عفونی: بررسی تعاملات میزبان–پاتوژن در HIV، سل و کووید-۱۹، که هم برنامههای دفاع ضدویروسی و هم اختلالات ایمنی ناشی از پاتوژن را آشکار کرده است.
۵. پزشکی دقیق (Precision Medicine)
یکی از چشماندازهای مهم scRNA-seq، کاربرد آن در پزشکی دقیق است.
-
با پروفایلبرداری از نمونههای بیماران در سطح تکسلول، میتوان امضاهای سلولی پیشبینیکننده پاسخ به درمان، پیشآگهی یا پیشرفت بیماری را شناسایی کرد.
-
در لوسمی، این روش توانسته است طبقهبندی دقیقتری نسبت به آسیبشناسی سنتی ارائه دهد و درمانهای هدفمندتری طراحی کند.
-
در تومورهای جامد، دادههای تکسلولی انتخاب اهداف ایمنیدرمانی و طراحی واکسنهای شخصیسازیشده سرطان را هدایت کرده است.
-
در بیماریهای خودایمنی، scRNA-seq برای تقسیمبندی بیماران بر اساس وضعیت ترنسکریپتومی سلولهای ایمنی پاتوژنیک به کار رفته است تا درمان متناسب با بیمار انتخاب شود.
۶. آینده فناوری
آینده scRNA-seq با ادغام روتین با سایر فناوریها روشنتر میشود:
-
ترکیب با فناوریهای فضایی برای مشخص کردن موقعیت سلولها در بافت و روابط عملکردی آنها.
-
ادغام با اپیژنومیک، پروتئومیک و متابولومیک برای دستیابی به یک دیدگاه جامع سامانهای از زیستشناسی سلول.
-
با افزایش ظرفیت و کاهش هزینه، scRNA-seq به ابزاری استاندارد در تحقیقات و حتی تشخیص بالینی تبدیل خواهد شد.
فصل ۸ – چالشها، محدودیتها و مسیرهای آیندهی scRNA-seq
تکنیک توالییابی RNA در تکسلول (scRNA-seq) انقلابی در درک ما از ناهمگنی سلولی ایجاد کرده و بینشهای بیسابقهای در مورد سیستمهای زیستی پیچیده فراهم آورده است. با این حال، با وجود تأثیر تحولآفرین آن، چالشها و محدودیتهای فنی و مفهومی متعددی همچنان مانع بهرهبرداری کامل از ظرفیت این فناوری هستند. غلبه بر این موانع برای بهبود کیفیت دادهها، تفسیرپذیری، و مقیاسپذیری ضروری است تا دامنهی کاربردهای زیستی و بالینی آن گسترش یابد.
۱. نویز و تغییرپذیری ذاتی دادههای تکسلولی
یکی از بزرگترین چالشها در scRNA-seq، مدیریت نویز فنی و تغییرپذیری ذاتی دادهها است. نویز فنی میتواند ناشی از عواملی مانند:
-
کارایی جذب RNA
-
سوگیریهای ناشی از تکثیر (amplification biases)
-
عمق توالییابی (sequencing depth)
این عوامل باعث ایجاد پدیدهای به نام Dropout میشوند، یعنی مواردی که یک ژن واقعاً بیان شده، اما در دادهها شناسایی نمیشود. این اختلالها تجزیهوتحلیلهای بعدی و تفسیر بیولوژیک را دشوار میکنند و نیازمند روشهای محاسباتی پیشرفته برای:
-
نرمالسازی (Normalization)
-
تخمین مقادیر از دسترفته (Imputation)
-
مدلسازی نویز
هستند. هرچند الگوریتمهای متعددی برای کاهش این اثرات توسعه یافتهاند، هنوز راهحل جهانی و واحدی وجود ندارد و همواره باید بین کاهش نویز و حفظ تغییرپذیری واقعی زیستی تعادل برقرار شود.
۲. مقیاسپذیری و مدیریت دادههای عظیم
با گسترش اندازه و پیچیدگی آزمایشهای scRNA-seq، موضوع مقیاسپذیری اهمیت بیشتری پیدا کرده است. پلتفرمهای مدرن قادرند پروفایل بیان ژن صدها هزار تا میلیونها سلول را ایجاد کنند که منجر به تولید دادههایی بسیار بزرگ میشود. این حجم عظیم داده، زیرساختهای محاسباتی فعلی را به چالش میکشد و نیازمند پیشرفت در:
-
طراحی الگوریتمهای کارآمد
-
پردازش موازی (Parallel computing)
-
راهحلهای مبتنی بر ابر (Cloud)
است.
علاوه بر این، ادغام دادههای چندوجهی (Multi-modal) — مثلاً ترکیب ترنسکریپتومیکس با اپیژنومیکس یا اطلاعات مکانی — لایههای جدیدی از پیچیدگی ایجاد میکند که نیازمند چارچوبهای تحلیلی نوآورانه برای تفسیر منسجم هستند.
۳. پیچیدگی محاسباتی تحلیلها
چالشهای محاسباتی فقط به اندازه دادهها محدود نمیشود؛ بلکه شامل شناسایی انواع سلولی، استنباط مسیرهای تمایز (Trajectory inference)، و بازسازی شبکههای تنظیم ژن نیز هست.
بسیاری از این وظایف بر تکنیکهای یادگیری بدوننظارت یا نیمهنظارتی متکی هستند که باید با دادههایی با ابعاد بالا و پراکندگی زیاد (Sparse) کار کنند.
نیاز است الگوریتمهای مقیاسپذیر و مقاوم توسعه یابند که با طرحهای آزمایشی و زمینههای بیولوژیک متنوع سازگار باشند. همچنین، افزایش تفسیرپذیری مدلهای محاسباتی اهمیت بالایی دارد تا یافتهها بتوانند به دانش زیستی کاربردی تبدیل شوند.
۴. راهکارهای نوظهور
پیشرفتهای نویدبخش در حال شکلگیریاند:
-
بهبود پروتکلهای آزمایشی برای کاهش نویز فنی و افزایش حساسیت (مثلاً با بهبود شیمی جذب RNA و فناوریهای توالییابی).
-
ادغام روشهای یادگیری ماشین و یادگیری عمیق برای مدلسازی وابستگیهای پیچیده و استخراج الگوهای معنادار از دادههای پرنویز.
-
توسعه مجموعهدادههای استاندارد و پروژههای جامعهمحور برای ایجاد خطوط تحلیلی بازتولیدپذیر و قابل مقایسه.
۵. آینده و مسیرهای توسعه
گسترش قابلیتهای scRNA-seq در آینده به پر کردن شکاف بین دادههای آزمایشی و بینش زیستی وابسته است. این شامل:
-
ادغام ترنسکریپتومیکس تکسلولی با اطلاعات مکانی و زمانی برای ثبت پویایی سلولها در محل اصلی آنها.
-
ترکیب scRNA-seq با لایههای دیگر اُمیکس برای دستیابی به دیدگاهی جامع از وضعیت سلولی.
-
توجه به مسائل اخلاقی، حریم خصوصی دادهها، و دسترسی برابر به فناوری بهویژه در مسیر استفاده بالینی.
✅ جمعبندی:
با اینکه scRNA-seq افقهای جدیدی را در زیستشناسی گشوده، چالشهای فنی و محدودیتهای محاسباتی هنوز نیازمند نوآوری و تلاشهای بینرشتهای هستند. غلبه بر این موانع میتواند به بینشهای عمیقتر و دقیقتر از پیچیدگی سلولی و نقش آن در سلامت و بیماری منجر شود.